

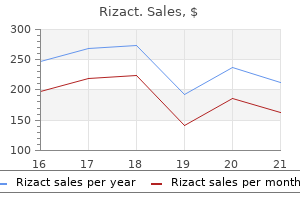
Rizact
Robert D. Stewart, MD, MPH
- Assistant Professor of Surgery
- Division of Cardiothoracic Surgery
- University of North Carolina School of Medicine
- Chapel Hill, North Carolina
Rizact dosages: 10 mg, 5 mg
Rizact packs: 4 pills, 8 pills, 12 pills, 24 pills, 32 pills, 48 pills

Discount 5 mg rizact with mastercard
Although less-well defined pain treatment for lumbar arthritis cheap rizact 5mg with visa, the overall gene and protein expression profiles of macrophages are additionally markedly influenced by the extracellular matrix sciatica pain treatment exercise rizact 5mg, hormones treatment for dog leg pain buy cheap rizact 5 mg online, and other immunomodulators, so that modified forms of irritation are related to macrophages current in lipid-rich environments, tumors, and metabolic ailments. Other antiinflammatory regulators of macrophages activation embrace glucocorticoids and prostaglandin E2. Although less-well outlined, the general gene and protein expressionCountway Medical Library profiles of macrophages are also markedly influenced by the extracellular matrix, hormones, and other immunomodulators, so that modified forms of irritation are Access Provided by: related to macrophages present in lipid-rich environments, tumors, and metabolic illnesses. Cell�cell interactions, in addition to intracellular regulatory networks, profoundly influence the functions of macrophages and are described in Chap. Alternative macrophage activation is important for survival throughout schistosomiasis and downmodulates T helper 1 responses and immunopathology. Imaging early macrophage differentiation, migration, and behaviors in stay zebrafish embryos. Macrophage and T cell dynamics through the growth and disintegration of mycobacterial granulomas. Cantharidin blisters: a method for investigating leukocyte trafficking and cytokine manufacturing at sites of irritation in people. Mouse macrophage hemagglutinin (sheep erythrocyte receptor) with specificity for sialylated glycoconjugates characterized by a monoclonal antibody. Expression and function of the sort 3 complement receptor in tissues of the creating mouse. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Transformation of mononuclear blood-cells into macrophages, epithelioid cells, and giant cells and large cells hanging-drop bloodcultures from lower vertebrates. Transformation of monocytes in tissue tradition into macrophages, epithelioid cells, and multinucleated large cells. Induction of multinucleated giant cell formation from human blood-derived monocytes by phorbol myristate acetate in in vitro tradition. Identification of macrophages and dendritic cells in the osteopetrotic (op/op) mouse. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating issue plus interleukin 4 and downregulated by tumor necrosis factor alpha. Activities of granulocyte-macrophage colony-stimulating factor revealed by gene switch and gene knockout studies. Phenotypic switching of adipose tissue macrophages with weight problems is generated by spatiotemporal variations in macrophage subtypes. Obesity-induced irritation: a metabolic dialogue within the language of irritation. Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy. Involvement of monocytes/macrophages as key elements within the growth and development of cardiovascular illnesses. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. Divalent cation-independent macrophage adhesion inhibited by monoclonal antibody to murine scavenger receptor. The mononuclear phagocyte system of the mouse outlined by immunohistochemical localization of antigen F4/80. Relationship between macrophages, Langerhans cells, reticular cells, and dendritic cells in lymphoid and hematopoietic organs. Macrophage activation and hemophagocytic syndrome in Langerhans cell histiocytosis: report of 30 cases. Fc chimeric protein containing the cysteine-rich area of the murine mannose receptor binds to macrophages from splenic marginal zone and lymph node subcapsular sinus and to germinal centers. Expression of mannose receptor and ligands for its cysteine-rich domain in venous sinuses of human spleen. Developmental regulation of sialoadhesin (sheep erythrocyte receptor), a macrophage-cell interaction molecule expressed in lymphohemopoietic tissues. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. The mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80: macrophages of endocrine organs. Differential charges of replacement of human dermal dendritic cells and macrophages during hematopoietic stem cell transplantation. Two distinct interstitial macrophage populations coexist throughout tissues in particular subtissular niches. Leukocyte adhesion molecules deficiency: its structural foundation, pathophysiology and implications for modulating the inflammatory response. Neurokinin-1 receptor: practical significance in the immune system in reference to selected infections and irritation. Full-length and truncated neurokinin-1 receptor expression and function throughout monocyte/macrophage22 / 23 �2021 McGraw Hill. Full-length and truncated neurokinin-1 receptor expression and performance throughout monocyte/macrophage differentiation. Differences in the size of the carboxyl terminus mediate practical properties of neurokinin-1 receptor. Development of a selected system for focusing on protein to metallophilic macrophages. Terms of Use � Privacy Policy � Notice � Accessibility Page 23 / 23 Countway Medical Library Access Provided by: Williams Hematology, 10e Chapter sixty nine: Classification and Clinical Manifestations of Disorders of Monocytes and Macrophages Marshall A. Monocytes are important sources for proinflammatory and inflammatory cytokines and, when inappropriately activated, may find yourself in the lymphohistiocytic hemophagocytic syndrome with fever, intravascular coagulation, and organ pathology. A variety of hematopoietic neoplasms may have a phenotype characterized by a large proportion of leukemic monocytes, promonocytes or monoblasts. Some circumstances of myelogenous leukemia have progenitor cells that mature preferentially into leukemic monocytes, including acute monoblastic or monocytic leukemia, continual myelomonocytic leukemia, and juvenile myelomonocytic leukemia. Two acquired diseases, bushy cell leukemia and aplastic anemia, end in a extreme melancholy of blood monocytes (along with different blood cell types). Inherited issues affecting white cells, corresponding to persistent granulomatous disease and Ch�diakHigashi syndrome, result in impaired monocyte operate. Monocyte dysfunction might accompany quite so much of severe sicknesses, such as sepsis, trauma, and most cancers. Monocytes additionally contribute to a selection of illnesses, such as Crohn illness and rheumatoid arthritis, by advantage of their being a principal supply of tumor necrosis factor. Monocytes play a pathogenetic role in other complex, acquired problems similar to thrombosis and atherogenesis. Macrophage or dendritic cell abnormalities principally involve tissues, where the diseases are referred to pathologically as histiocytosis. These issues can be inherited, corresponding to familial hemophagocytic lymphohistiocytosis; inflammatory, similar to infectious hemophagocytic lymphohistiocytic syndrome; or clonal (neoplastic), corresponding to Langerhans cell histiocytosis. They may finish up from an inherited enzyme insufficiency in macrophages that result in exaggerated storage of macromolecules and subsequent tissue injury, such as in Gaucher illness.
Generic 5 mg rizact
In sufferers with delicate disease pain & depression treatment cheap rizact 10 mg on-line, the serum indirect bilirubin peaks by the fourth or fifth day after which declines slowly advanced diagnostic pain treatment center new haven buy rizact 10 mg amex. Premature infants may have greater levels of serum bilirubin for an extended duration due to decrease hepatic glucuronyl transferase activity pain treatment center west plains mo cheap 5 mg rizact fast delivery. With rising severity, the toddler develops a high-pitched cry, fever, hypertonia progressing to frank opisthotonos, and irregular respiration. The infants then develop any or all the classic sequelae of kernicterus: choreoathetoid cerebral palsy, gaze abnormalities, especially in upward gaze, sensorineural hearing loss, and cognitive deficits. Abnormal or absent brainstem auditory evoked potentials and magnetic resonance imaging scans demonstrating the attribute bilateral lesions of the globus pallidus help verify the scientific analysis of kernicterus. Terms of Use � Privacy Policy � Notice � Accessibility during energetic hemolysis are hypothesized to inhibit bilirubin�albumin binding. Alternatively, many circumstances that potentially compromise the blood� brain barrier, such as prematurity, acidosis, hypoxemia, hypothermia, and hypoglycemia, are present in severely affected infants, making them more encephalopathy is initially marked by lethargy, poor feeding, and hypotonia. With growing severity, the toddler develops a high-pitched cry, fever, Countway Medical Library hypertonia progressing to frank opisthotonos, and irregular respiration. The infants then develop any or all of the classic sequelae of kernicterus: Access Provided by: choreoathetoid cerebral palsy, gaze abnormalities, particularly in upward gaze, sensorineural hearing loss, and cognitive deficits. The scientific presentation of bilirubin encephalopathy in preterm infants could additionally be less distinctive. Alternatively, many situations that doubtlessly compromise the blood� brain barrier, such as prematurity, acidosis, hypoxemia, hypothermia, and hypoglycemia, are present in severely affected infants, making them more weak to bilirubin encephalopathy. Hydrops recurs in 90% of affected pregnancies, usually at an earlier gestation in subsequent pregnancies. The history of prior blood transfusions could additionally be obtained in girls sensitized to antigens other than RhD, especially if Kell alloimmunization is detected. Establishment of paternity for each pregnancy is especially related in each Rh and Kell alloimmunization as a end result of the fetus is at risk provided that the daddy is positive for the antigen in query. Nonimmune causes account for roughly 85% of all affected individuals, and mostly end result from cardiac (20%), hematologic (10%), chromosomal (9%), thoracic (2%), gastrointestinal (1%), or urinary tract (1%) anomalies; lymphatic dysplasia (15%); intrauterine infections (7%); twin-to-twin transfusion syndrome (4%); and inborn errors of metabolism (1%) disorders. Other rarer congenital, placental, and idiopathic causes can also result in hydrops fetalis. Maternal parvovirus B19 infection at any time throughout gestation may cause nonimmune hydrops, profound fetal anemia, and death. Hepatitis or obstructive biliary illnesses present with direct hyperbilirubinemia, most often after the first week of life. Maternal parvovirus Countway Medical Library B19 infection at any time during gestation can cause nonimmune hydrops, profound fetal anemia, and dying. Amino acid substitutions situated in extracellular domains lead to totally different forms of partial D phenotype. Amino acid substitutions located in the transmembrane or intracellular segments of the RhD protein most frequently end in a weak D phenotype. The expressed RhD antigen is most frequently decreased quantitatively however not qualitatively, so carriers are usually not prone to anti-D immunization. Women whose genotyping returns as being weak D varieties 1, 2, or three could be treated as being RhD positive for transfusion and pregnancy functions. Women whose genotyping returns with any other weak D or partial D variant should be treated as RhD adverse for transfusion and being pregnant purposes. Until genotyping info is obtained, girls with potential weak or partial RhD must be treated as being RhD negative and, if indicated, treated with anti-D immune globulin prophylaxis. RhD-negative pregnant women who appear to have each anti-D and anti-C, especially if at an analogous strengths of reactivity serologically, require special consideration. In these conditions, the laboratory should decide whether the antibodies are really anti-D/anti-C rather than an anti-G as a result of a patient who develops anti-C and/or anti-G antibodies but not anti-D ought to obtain anti-D immune globulin prophylaxis. Terms of Use � Privacy Policy � Notice � Accessibility such as chorionic villus sampling, amniocentesis, therapeutic abortion, exterior cephalic model, cesarean section, and guide removal of the placenta, and from pathologic circumstances similar to abdominal trauma, spontaneous abortion, or ectopic pregnancy. The average quantity of fetal blood in the maternal circulation after delivery is roughly zero. Subsequently, approximately 5�15 weeks after exposure to the RhD-positive purple cells, antiD IgG antibodies capable of crossing the placenta are produced. Memory T and B cells which are generated after the preliminary immune response are lengthy lived, and exposure to the antigen even years later ends in an accelerated antibody response because of rapid proliferation of antigen-specific clones. Any alloantibody capable of inducing hemolysis or suppressing erythropoiesis may be clinically important to growing fetuses. However, the mere presence of antibodies on screening exams will not be clinically vital due to the distinctive characteristics of some antibodies. Clinical observations of inappropriately low ranges of circulating reticulocytes and normoblasts for the degree of anemia have lengthy been noted in affected fetuses, and suppression of erythropoiesis has been established by in vitro studies exhibiting that growth of Kell-positive erythroid progenitor cells is inhibited by monoclonal IgG and IgM anti-Kell antibodies. Antigen copy number, in addition to different characteristics of the antigens or antibodies (potentially together with IgG subtype), may impact the clinical significance of the alloantibodies. Terms of Use � Privacy Policy � Notice � Accessibility occurring antibodies against the A and B antigens that are of the IgG class and are thus capable of crossing the placenta. A recurrence price of 88% has been reported in siblings having the same blood kind because the affected index child, with two-thirds of the affected siblings requiring remedy. Terms of Use � Privacy Policy � Notice � Accessibility Exchange transfusion frequency Approximately two or three No Occasional Page eight / 30 been reported in siblings having the same blood sort as the affected index child, with two-thirds of the affected siblings requiring therapy. Terms of Use � Privacy Policy � Notice � Accessibility Page 9 / 30 Algorithm for the medical management of an alloimmunized being pregnant. The child of an antigen-negative mom and a heterozygous antigenpositive father has a 50% likelihood of being antigen optimistic and thus being affected by maternal allosensitization. When the father is heterozygous or paternal zygosity is unknown, dedication of fetal blood type early in pregnancy allows early establishment of monitoring and therapy in antigenpositive fetuses which would possibly be at risk while forestalling invasive and potentially dangerous procedures in antigen-negative fetuses. These embody blood obtained by cordocentesis, chorionic villus sampling, and cervical tissue obtained by transvaginal lavage. The practice pointers and proposals for pregnancy-associated immunohematologic and molecular testing have been established within the United States by the American Association of Blood Banks. These maternal samples must also be screened for the presence of red cell alloantibodies. Luban performed in parallel with beforehand frozen samples to reduce the possibility that modifications within the titer end result from differences in method or11 / 30 �2021 McGraw Hill. Terms of Use � Privacy Policy � Notice � Accessibility 87,89 A crucial titer is outlined as the titer associated with significant threat of fetal anemia or hydrops and is the edge at reagent pink cell choice. Testing should ideally be performed in parallel with beforehand frozen samples to reduce the chance that adjustments within the titer outcome from variations in technique or reagent pink cell choice. When the critical titer is reached and a choice is made to monitor the fetus by ultrasonography or amniocentesis, additional antibody titration performs no function in evaluation of fetal status.
Syndromes
- Television viewing has been associated with higher rates of attention problems in children.
- Ill-fitting dentures
- Bright red vaginal bleeding
- Never leave children or pets unattended in a car in the sun -- even for a few minutes.
- Pancreatitis (swollen and inflamed pancreas)
- Get worse with coughing or exercise, or with a change in body position
Buy generic rizact 5 mg
Increased concentrations of regular neutrophils per se are usually not associated with clinical manifestations pain management treatment guidelines safe rizact 5 mg, though increased concentrations of leukemic neutrophil precursors can produce clinical manifestations of microcirculatory leukostasis (Chap pain treatment centers of america carl covey discount rizact 10 mg mastercard. Neutrophils additionally play a job in deleterious vascular or tissue results alpha pain treatment center berwyn il cheap 5mg rizact mastercard, as famous within the final entries in Table 62�1 (see "Neutrophilia" later). If the neutrophil count drops further, the chance of an infection could increase if the decrease displays a lower in flux fee into the tissues. The relationship of frequency or type of an infection to neutrophil concentration is imperfect. The explanation for the neutropenia, the coincidence of monocytopenia or lymphopenia, concurrent use of alcohol or glucocorticoids, publicity to nosocomial infections, and other elements influence the probability of an infection. A breakdown within the barrier perform of the pores and skin or circumstances corresponding to indwelling catheters additionally improve the chance of infection in severely neutropenic topics. However, any site can turn into infected, and gram-negative organisms, viruses, or opportunistic organisms may be concerned. If the neutrophil depend approaches zero (agranulocytosis), high fever; chills; necrotizing, painful oral ulcers (agranulocytic angina); and prostration might happen, presumably on account of sepsis. In the preantibiotic era, persistent agranulocytosis had a fatality rate approaching 100%. Even with bactericidal, broad-spectrum antibiotics, severe, sustained neutropenia or agranulocytosis is a serious illness with a excessive fatality rate. For example, lack of pneumonic consolidation is attribute of pneumonia in granulocytopenic topics. An exudate, swelling, heat, and regional adenopathy are much much less prevalent in granulocytopenic patients. Fever is common, and native pain, tenderness, and erythema nearly at all times are current despite a marked reduction in neutrophils. Chronic idiopathic (benign) neutropenia is related to obvious normal granulopoiesis within the marrow and is asymptomatic even when the neutropenia has been current for extended intervals, sometimes within the face of neutrophil counts approaching zero for prolonged periods. Monocyte counts are normal, which may help in host defenses because monocytes are effective phagocytes. Chronic idiopathic (symptomatic) neutropenia often is associated with pyoderma and otitis media in youngsters. The former often is brought on by Staphylococcus aureus, Escherichia coli, and Pseudomonas spp. Pneumonia, lung abscesses, stomatitis, hepatic abscesses, or infections in different sites can occur. Chronic cyclic neutropenia is characterized by periodic oscillations in the variety of neutrophils, with the nadir occurring at approximately 3-week intervals. Terms of Use � Privacy Policy � Notice � Accessibility Some individuals have neutropenia because a bigger fraction of their blood neutrophils is within the marginal somewhat than the circulating pool. Countway Medical Library Access Provided by: Chronic cyclic neutropenia is characterised by periodic oscillations within the number of neutrophils, with the nadir occurring at roughly 3-week intervals. Furuncles, carbuncles, cellulitis, infected cuts with lymphangitis, continual gingivitis, and abscesses of the axilla or groin may occur. Although extreme infections may be deadly, life-threatening issues are uncommon. The biking entails other hematopoietic cells as properly, but the neutropenia is probably the most consequential functionally (Chap. Some people have neutropenia as a outcome of a larger fraction of their blood neutrophils is within the marginal quite than the circulating pool. Certain medicine, similar to glucocorticoids or hematopoietic progress factors and minocycline, can induce neutrophilia, as can ethylene glycol intoxication (see Table 62�1). Neutrophilia exceeding 50 � 109 neutrophils/L has been designated a "leukemoid response," if not a myeloproliferative neoplasm, and displays an underlying inflammatory (eg, pancreatitis), infectious (eg, pneumococcal pneumonia), or neoplastic (eg, carcinoma of the lung) trigger. Chronic granulomatous disease132,133 and Ch�diak-Higashi disease161,162 are two examples of inherited defects. Among the acquired problems are those extrinsic to the cell, as within the motion, chemotactic, or phagocytic defects of diabetes mellitus, the results of alcohol abuse, or glucocorticoid extra. Acquired intrinsic issues usually are manifestations of clonal hematopoietic (myeloid) disorders such as acute myelogenous leukemia (Chap. Severe defects in bacterial killing, as occur in persistent granulomatous disease, end in S. Suppurative lymphadenitis, pneumonia, dermatitis, hepatic abscesses, osteomyelitis, and stomatitis happen, and continual granulomatous reactions in these websites give the illness its name. Mild functional disorders predispose to infections that happen infrequently and reply readily to antibiotics. Neutrophils, nevertheless, can transiently occlude capillaries, as decided by supravital microscopy, and such occlusions could cut back native blood move transiently and contribute to the development of ischemia. Impairment of reperfusion of the coronary microcirculation has been thought to be dependent, in part, on neutrophil plugging of myocardial capillaries, but these results can occur at regular neutrophil concentrations. Terms of Use � Privacy Policy � Notice � Accessibility 98 episodes and the severity of continual atherosclerosis. Neutrophils, however, can transiently occlude capillaries, as Countway Medical Library determined by supravital microscopy, and such occlusions may cut back native blood circulate transiently and contribute to the event of ischemia. Access Provided by: Impairment of reperfusion of the coronary microcirculation has been thought to be dependent, partly, on neutrophil plugging of myocardial capillaries, however these effects can occur at regular neutrophil concentrations. An elevated neutrophil depend is a characteristic of sickle cell illness and is a unfavorable prognostic variable, rising the chance of vasoocclusive events. Neutrophil adhesion to the vascular wall is an intrinsic a half of the vasoocclusive events and the salutary effect of hydroxyurea is related partially to the decrease in neutrophil concentration that accompanies its use. Diabetic retinopathy has been ascribed in part to the consequences of hyperadhesive neutrophils on retinal capillaries. This motion may clarify, for example, the event of carcinoma of the bowel in patients with persistent ulcerative colitis and the relationship between elevated leukocyte count and the prevalence of lung cancer, unbiased of the effect of cigarette usage. The oxidants, especially hypochlorous acid and chloramines, released by the neutrophil are extraordinarily brief lived and will play a role in tissue harm by inactivating a number of protease inhibitors in tissue fluids, permitting proteases, particularly elastase, collagenase, and gelatinase, to trigger tissue injury. Notably, the ratio has been proven to be superior to neutrophil counts as a predictor of bacteremia in sufferers admitted to emergency departments or intensive care models. With an abnormal temperature, both above or under normal, an elevated respiratory rate and heart price, the constellation of findings method a probability of 90% that sepsis is current. Genetics and pathophysiology of severe congenital neutropenia syndromes unrelated to neutrophil elastase. Recent advances in the understanding of genetic defects of neutrophil quantity and performance. Recent advances in understanding the pathogenesis and management of reticular dysgenesis. Recent advances in the pathogenesis and remedy of nonimmune neutropenias within the neonate.

Buy rizact 5 mg amex
Posttraumatic epilepsy and acute intermittent porphyria: effects of phenytoin pain treatment center seattle wa generic rizact 5mg, carbamazepine arizona pain treatment center phoenix az rizact 10 mg overnight delivery, and clonazepam pain treatment for ra cheap rizact 5mg line. Treatment choices in acute porphyria, porphyria cutanea tarda, and erythropoietic protoporphyria. Terms of Use � Privacy Policy � Notice � Accessibility hemoproteins and regulatory heme pools. Intravenous heme-albumin in acute intermittent porphyria: evidence for repletion of hepatic 249. Intravenous heme-albumin in acute intermittent porphyria: proof for repletion of hepatic hemoproteins and regulatory heme swimming pools. Circulatory collapse associated with hemin remedy for acute intermittent porphyria. Transitory renal failure following rapid administration of a comparatively large amount of hematin in a affected person with acute intermittent porphyria in scientific remission. Prevention of premenstrual exacerbation of hereditary coproporphyria by gonadotropin-releasing hormone analogue. An audit of using gonadorelin analogues to forestall recurrent acute signs in sufferers with acute porphyria within the United Kingdom. The medical and biochemical features of variegate porphyria: an analysis of 300 cases studied at Groote Schuur Hospital, Cape Town. Biochemical and genetic characterization of four instances of hereditary coproporphyria in Spain. The Human Gene Mutation Database: in course of a comprehensive repository of inherited mutation data for medical analysis, genetic prognosis and next-generation sequencing studies. The Human Gene Mutation Database: towards a complete repository of inherited mutation information for Access Provided by: medical research, genetic analysis and next-generation sequencing studies. Plasma fluorescence scanning and fecal porphyrin evaluation for the diagnosis of variegate porphyria: exact determination of sensitivity and specificity with detection of protoporphyrinogen oxidase mutations as a reference standard. Neonatal hemolytic anemia due to inherited harderoporphyria: medical traits and molecular basis. Immunoreactive uroporphyrinogen decarboxylase within the liver in porphyria cutanea tarda. Effect of iron and ascorbate on uroporphyria in ascorbate-requiring mice as a mannequin for porphyria cutanea tarda. Circulating pro- and antioxidant components in iron and porphyrin metabolism disorders. Functional penalties of naturally occurring mutations in human uroporphyrinogen decarboxylase. A retrospective evaluation of 17 cases from a single centre and review of the literature. Complement cleavage products within the phototoxic reaction of porphyria cutanea tarda. Transdermal estrogen alternative remedy in postmenopausal ladies previously handled for porphyria cutanea tarda. Hereditary uroporphyrinogen-decarboxylase deficiency predisposing porphyria cutanea tarda (chronic hepatic porphyria) in females after oral contraceptive medicine. Effect of estradiol on the induction of porphyria by hexachlorobenzene within the rat. Patients with persistent hepatitis C attaining a sustained virological response to peginterferon and ribavirin therapy recuperate from impaired hepcidin secretion. The frequency of hemochromatosis-associated alleles is elevated in British sufferers with sporadic porphyria cutanea tarda. Co-inheritance of mutations in the uroporphyrinogen decarboxylase and hemochromatosis genes accelerates the onset of porphyria cutanea tarda. Hypertrichosis because of porphyria cutanea tarda related to blastic transformation of myelofibrosis. Erythropoietin for the therapy of porphyria cutanea tarda in a patient on long-term hemodialysis. Recovery from porphyria cutanea tarda with no particular therapy other than avoidance of hepatic toxins. Childhood-onset porphyria cutanea tarda: successful remedy with low-dose hydroxychloroquine (Plaquenil). Studies on low dose chloroquine therapy and the motion of chloroquine in symptomatic porphyria. Low-dose hydroxychloroquine is as effective as phlebotomy in therapy of sufferers with porphyria cutanea tarda. Complex formation between chloroquine and ferrihaemic acid in vitro, and its impact on the antimalarial motion of chloroquine. Management of porphyria cutanea tarda within the setting of persistent renal failure: a case report and review. Haemodialysis-related porphyria cutanea tarda and therapy by recombinant human erythropoietin. Removal of plasma porphyrins with high-flux hemodialysis in porphyria cutanea tarda associated with end-stage renal disease. Porphyria cutanea tarda within the setting of renal failure: response to renal transplantation. Correction of uroporphyrinogen decarboxylase deficiency (hepatoerythropoietic porphyria) in Epstein-Barr virus-transformed B- cell strains by retrovirus-mediated gene switch: fluorescence-based selection of transduced cells. Terms of Use � Privacy Policy � Notice � Accessibility Page fifty one / fifty one Countway Medical Library Access Provided by: Williams Hematology, 10e Chapter 60: Polyclonal and Hereditary Sideroblastic Anemias Prem Ponka; Amel Hamdi; Josef T. These cells are erythroid precursors that have accrued irregular amounts of mitochondrial iron. A number of abnormalities of porphyrin metabolism in affected erythroid cells have been documented. Hereditary sideroblastic anemias are often X-linked, as the result of mutations within the erythroid type of 5-aminolevulinic acid synthase. Acquired sideroblastic anemias can occur because of copper deficiency or the ingestion of medicine, alcohol, or toxins similar to lead or zinc. Patients with acquired sideroblastic macrocytic anemia and variable levels of thrombocytopenia and leukopenia caused by copper deficiency have been recognized more regularly; the hematologic abnormalities usually resolve after copper alternative. Ring sideroblasts are also a feature of myelodysplastic neoplasms, which are mentioned in Chap. Some patients with sideroblastic anemia could respond to pharmacologic doses of pyridoxine. Iron loading is frequent in the sideroblastic anemias and can be treated by phlebotomy when the anemia is mild or with iron chelators (Chap. Chronic neoplastic disease Page 1 / 18 Sideroblastic anemias are a heterogeneous group of problems which have as a typical characteristic the presence of (a) giant numbers of pathological Countway Medical Library sideroblasts in the marrow, which characteristically display irregular mitochondrial iron accumulation in a circumnuclear position, the location of Access Provided by: mitochondria in erythroblasts; these are referred to as ring sideroblasts; (b) ineffective erythropoiesis; (c) increased ranges of tissue iron; and (d) varying proportions of hypochromic erythrocytes in the blood. Heteroplasmic point mutations in subunit 1 of the mitochondrial cytochrome oxidase gene 3. Acquired polyclonal sideroblastic anemia can also develop as a result of the administration of certain drugs, publicity to toxins, or coincident to neoplastic or inflammatory illness.
Generic 10mg rizact with visa
Risk Stratification for Patients with Essential Thrombocythemia No High-Risk Features High Risk Low Risk Age >60 years Prior thrombosis Platelets >1500 � 109/L Age <40 years Intermediate Risk Age 40�60 years Once cytoreductive remedy is instituted hip pain treatment for dogs discount rizact 10mg, dose adjustment is beneficial to keep the platelet and leucocyte counts within the normal vary pain management service dogs quality 5 mg rizact. Hydroxyurea treatment for lingering shingles pain buy 5mg rizact visa, a ribonucleotide reductase inhibitor also identified as hydroxycarbamide, is extensively considered first-line remedy for patients requiring treatment, and is the one cytoreductive agent confirmed to scale back thrombotic occasions in a randomized, controlled trial. Terms of Use � Privacy Policy � Notice � Accessibility Page 11 / 21 multiple cytotoxic brokers, lack of correct controls, retrospective data assortment, and comparatively brief follow-up. Anagrelide, a quinazoline derivative, reduces the platelet depend by inhibition of megakaryocyte differentiation. Although the white cell rely is unaffected, anemia is widespread and often progressive. Use of this drug requires particular warning in older adult sufferers or those with preexisting cardiac illness. In this study, anagrelide-treated sufferers experienced decreased event-free survival (P =. Terms of Use � Privacy Policy � Notice � Accessibility Follow-up 730 patient-years 2653 patient-years Page 12 / 21 as second-line therapy for sufferers in whom hydroxyurea remedy is inadequate or not tolerated. In addition, the ethical foundation for performing noninferiority trials may be questioned. Treatment is commonly related to significant unwanted facet effects, together with flulike signs and psychiatric disturbance that may mandate cessation of remedy. Because this agent is free from leukemogenic or teratogenic results,84 interferon- is commonly used for youthful sufferers or throughout conception and being pregnant. Pegylated interferon-, for which much less frequent administration is required, could also be more convenient however the facet effect profile seems much like the native compound. Radioactive (32P) phosphorus and alkylating brokers similar to busulfan are efficient at controlling the platelet rely however are related to an elevated risk of development to acute leukemia, particularly when used sequentially with hydroxyurea, and thus ought to be prevented in all but very uncommon patients. In this paradigm, very-low-risk patients have not considered one of the three threat components, and may be appropriate for observation alone or, at most, low-dose aspirin remedy. High-risk sufferers display all three danger elements and, depending on the nature of their prior thrombosis (arterial or venous), must be treated with hydroxyurea and either twice-daily aspirin (arterial thrombosis) or hydroxyurea and systemic anticoagulation (eg, low-dose heparin or a direct thrombin inhibitor). Whether the usage of aspirin or cytoreductive agents can improve pregnancy end result is uncertain, with research reporting contradictory outcomes. Anagrelide can cross the placenta with unknown results on fetal growth and should also be averted. Interferon- is nonteratogenic84 and is the agent of alternative for sufferers with high-risk disease should cytoreductive remedy be required during pregnancy. In animal studies, hydroxyurea is related to decreased spermatogenesis and genetic injury to spermatogonia. In general, antiplatelet brokers must be stopped 7�10 days earlier than major surgical procedure or surgery to critical websites, and recommenced as soon as the surgeon is confident of secure hemostasis. Postoperative thromboprophylaxis ought to be administered based on local protocols. For sufferers receiving cytoreductive remedy, control of blood counts should be optimized preoperatively and interruptions in therapy stored to a minimum. Thromboprophylaxis and day by day monitoring of blood counts is recommended during the postoperative interval. Data from cancer registries and retrospective studies point out that general survival is modestly reduced compared with inhabitants controls. However, administration paradigms have modified considerably over the past 10�20 years, together with extra aggressive intervention to prevent thrombosis and decreasing use of leukemogenic agents. Terms of Use � Privacy Policy � Notice � Accessibility finest established of that are age larger than 60 years or a historical past of previous thrombosis. Data from cancer registries and retrospective studies point out that Countway Medical Library Access Provided by: total survival is modestly reduced in contrast with population controls. These adjustments, which can be associated to observed improvements in affected person outcomes in latest times,93 render some long-term follow-up studies troublesome to interpret. As mentioned above (see "Cytoreductive Therapy") numerous predictive factors for thrombotic problems have been recognized (Table 84�6), the most effective established of which are age higher than 60 years or a historical past of earlier thrombosis. The incidence of each problems will increase progressively with disease duration. Choice of therapy also performs a task, with anagrelide growing the risk of myelofibrotic transformation in contrast with hydroxyurea30 and genotoxic agents growing the danger of leukemia, particularly when used sequentially with hydroxyurea. Myelofibrosis with myeloid metaplasia in survivors of the atomic bomb in Hiroshima. Evidence that important thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Population-based incidence and survival figures in important thrombocythemia and agnogenic myeloid metaplasia: an Olmsted County Study, 1976-1995. An amphipathic motif at the transmembrane-cytoplasmic junction prevents autonomous activation of the thrombopoietin receptor. Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic-defective pathogenic TpoR mutants. Whole-exome sequencing of polycythemia vera revealed novel driver genes and somatic mutation shared by T cells and granulocytes. Risk for arterial and venous thrombosis in patients with myeloproliferative neoplasms: a population-based cohort research Ann Intern Med. Erythromelalgic, thrombotic and hemorrhagic manifestations in 50 cases of thrombocythemia. Prognostic components for thrombosis, myelofibrosis, and leukemia in important thrombocythemia: a research of 605 patients. Association of V617F Jak2 mutation with the risk of thrombosis among patients with essential thrombocythaemia or idiopathic myelofibrosis: a systematic evaluation. Reticulin accumulation in essential thrombocythemia: prognostic significance and relationship to therapy. Factors associated to the development of acquired von Willebrand syndrome in sufferers with �2021 McGraw Hill. Terms of Use � Privacy Policy � Notice � Accessibility essential thrombocythemia and polycythemia vera. Reticulin accumulation in important thrombocythemia: prognostic significanceCountway Medical Library and relationship to therapy. Factors related to the event of acquired von Willebrand syndrome in patients with essential thrombocythemia and polycythemia vera. Essential thrombocythemia past the first decade: life expectancy, long-term complication rates, and prognostic components. Bone marrow pathology in important thrombocythemia: interobserver reliability and utility for identifying disease subtypes. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Long-term incidence of hematological evolution in three French potential studies of hydroxyurea and pipobroman in polycythemia vera and important thrombocythemia. Increased incidence of acute leukemia in polycythemia vera associated with chlorambucil remedy.
Order rizact 10mg on-line
However shoulder pain treatment yahoo 5 mg rizact overnight delivery, etoposide pain treatment guidelines rizact 5 mg visa, which has particular activity against activated T cells pain treatment center franklin tn generic rizact 10mg without prescription, could also be necessary to control pathologic irritation. Terms of Use � Privacy Policy � Notice � Accessibility Supportive Care etoposide/dexamethasone/cyclosporine whose illness had relapsed whereas on remedy. Transplant recipients are at excessive danger for both transplant associated mortality and graft rejection. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Dendritic cells, monocytes and macrophages: a unified nomenclature primarily based on ontogeny. Origin, homeostasis and performance of Langerhans cells and different langerin-expressing dendritic cells. Pathological consequence of misguided dendritic cell differentiation in histiocytic ailments. Histiocytosis X (eosinophilic granuloma of bone, Letterer-Siwe illness, and Schueller-Christian disease). Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000-2004. Ethnicity, race, and socioeconomic standing influence incidence of Langerhans cell histiocytosis. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile in contrast with epidermal Langerhans cells. Blood-derived dermal langerin+ dendritic cells survey the pores and skin in the steady state. Langerhans cell histiocytoisis: present insights in a molecular age with emphasis on medical oral and maxillofacial pathology follow. Thymus and mediastinal node involvement in childhood Langerhans cell histiocytosis: long-term follow-up from the French national cohort. Histologic patterns of thymic involvement in Langerhans cell proliferations: a clinicopathologic study and evaluation of the literature. Pulmonary involvement in pediatric-onset multisystem Langerhans cell histiocytosis: effect on target and outcome. Macrophage activation and hemophagocytic syndrome in langerhans cell histiocytosis: report of 30 circumstances. Gastrointestinal tract involvement in Langerhans cell histiocytosis: case report and literature evaluate. A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Cladribine and cytarabine in refractory multisystem Langerhans cell histiocytosis: outcomes of a world phase 2 research. The relation of Langerhans cell histiocytosis to acute leukemia, lymphomas, and other stable tumors. Cast and brace treatment of eosinophilic granuloma of the spine: long-term follow-up. Efficacy of vinblastine in central nervous system Langerhans cell histiocytosis: a nationwide retrospective examine. Neurodegenerative central nervous system Langerhans cell histiocytosis and coincident hydrocephalus handled with vincristine/cytosine arabinoside. Clofarabine salvage therapy in refractory multifocal histiocytic problems, together with Langerhans cell histiocytosis, juvenile xanthogranuloma and Rosai-Dorfman illness. Terms of Use � Privacy Policy � Notice � Accessibility intravenous immunoglobulin and chemotherapy. Clofarabine salvage remedy in refractory multifocal histiocytic issues, including Langerhans cell Access Provided by: histiocytosis, juvenile xanthogranuloma and Rosai-Dorfman disease. Retinoic acid remedy in "degenerative-like" neuro-langerhans cell histiocytosis: a potential pilot examine. Excellent remission rates with limited toxicity in relapsed/refractory Langerhans cell histiocytosis with pulse dexamethasone and lenalidomide in children. Cytosine-arabinoside, vincristine, and prednisolone within the therapy of children with disseminated Langerhans cell histiocytosis with organ dysfunction: expertise at a single establishment. Clofarabine salvage therapy for refractory high-risk langerhans cell histiocytosis. Vemurafenib for refractory multisystem Langerhans Cell Histiocytosis in kids: a world observational research. Haematopoietic stem cell transplantation for refractory Langerhans cell histiocytosis: consequence by intensity of conditioning. Incidence of growth hormone deficiency in pediatric-onset Langerhans cell histiocytosis: efficacy and security of progress hormone remedy. Permanent penalties in Langerhans cell histiocytosis sufferers: a pilot study from the Histiocyte SocietyLate Effects Study Group. Cognitive consequence of long-term survivors of multisystem langerhans cell histiocytosis: a singleinstitution, cross-sectional research. Central nervous system-related everlasting penalties in sufferers with Langerhans cell histiocytosis. Evaluation and therapy of Langerhans cell histiocytosis patients with central nervous system abnormalities: current views and new vistas. Clonal relationship between precursor T-lymphoblastic leukaemia/lymphoma and Langerhans-cell histiocytosis. High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis. Adult disseminated Langerhans cell histiocytosis: incidence, racial disparities and long-term outcomes. Systemic histiocytosis (Langerhans cell histiocytosis, Erdheim-Chester illness, Destombes-RosaiDorfman disease): from oncogenic mutations to inflammatory disorders. Evidence that Langerhans cells in grownup pulmonary Langerhans cell histiocytosis are mature dendritic cells: importance of the cytokine microenvironment. Is high-resolution computed tomography a reliable software to predict the histopathological activity of pulmonary Langerhans cell histiocytosis Vinblastine chemotherapy in grownup sufferers with langerhans cell histiocytosis: a multicenter retrospective examine. Management of adult sufferers with Langerhans cell histiocytosis: suggestions from an expert panel on behalf of Euro-Histio-Net. Outcome after radiation remedy for Langerhans cell histiocytosis depends on site of involvement. Methylphenidate for the remedy of depressive signs, including fatigue and apathy, in medically sick older adults and terminally unwell adults.
Pepperrot (Horseradish). Rizact.
- Are there any interactions with medications?
- Dosing considerations for Horseradish.
- What is Horseradish?
- Urinary tract problems, fluid retention (edema), cough, bronchitis, achy joints and muscles, gout, gallbladder disorders, sciatic nerve pain, colic, intestinal worms in children, and other conditions.
- Are there safety concerns?
- How does Horseradish work?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96281

Quality rizact 10 mg
Changes in plasma and extracellular fluid volumes in sufferers with essential hypertension during long-term therapy with Countway Medical Library hydrochlorothiazide cape fear pain treatment center cheap rizact 5 mg free shipping. Personal communication and direct expertise with about 100 affected subjects; 2009 pain treatment center of the bluegrass lexington ky generic rizact 10mg otc. Familial erythrocytosis associated with a short deletion within the erythropoietin receptor gene pain treatment center sawgrass quality 5mg rizact. Higher offspring survival among Tibetan girls with excessive oxygen saturation genotypes residing at four,000 m. Oxygen saturation will increase during childhood and decreases during maturity among excessive altitude native Tibetians residing at 3,8004,200m. Effects of polycythemia on systemic endothelial function in chronic hypoxic lung illness. Cardial decompensation caused by hypertension and polyglobulia associated with multiple renal oncocytomas. Erythropoiesis after kidney transplantation: the position of erythropoietin, burst selling exercise and early erythroid progenitor cells. Endothelin-1, vascular endothelial progress factor and systolic pulmonary artery pressure in patients with Chuvash polycythemia. Comparison of molecular markers in a cohort of patients with continual myeloproliferative issues. In vitro erythropoiesis in polycythemia vera and different myeloproliferative disorders. Identification of latent myeloproliferative illness in patients with Budd-Chiari syndrome utilizing Xchromosome inactivation patterns and in vitro erythroid colony formation. Spontaneous erythroid colony formation as the clue to an underlying myeloproliferative disorder in patients with Budd-Chiari syndrome or portal vein thrombosis. In vitro colony tradition and chromosomal studies in hepatic and portal vein thrombosis- possible proof of an occult myeloproliferative state. Serum erythropoietin within the analysis of polycythaemia and after phlebotomy therapy. Identification of masked polycythemia vera from sufferers with idiopathic marked thrombocytosis by endogenous erythroid colony assay. Pathogenetic mechanisms of polycythemia vera and congenital polycythemic issues. Transcriptional analysis of the energetic X-chromosome in regular and clonal hematopoiesis. Clonal stability of blood cell lineages indicated by X-chromosomal transcriptional polymorphism. Nonrandom X-inactivation patterns in regular females: lyonization ratios differ with age. Clonal haemopoiesis in normal aged girls: implications for the myeloproliferative disorders and myelodysplastic syndromes. Acquired skewing of X-chromosome inactivation patterns in myeloid cells of the aged suggests stochastic clonal loss with age. Clonal hematopoiesis in familial polycythemia vera suggests the involvement of multiple mutational occasions Page 37 / 38, Josef T. Angiotensin-converting-enzyme inhibition remedy in altitude polycythaemia: a potential randomised trial. Congenital erythrocytosis with elevated erythropoietin stage: an incorrectly set "erythrostat" Effects of phlebotomy on a affected person with secondary polycythemia and angina pectoris. Terms of Use � Privacy Policy � Notice � Accessibility Page 38 / 38 Countway Medical Library Access Provided by: Williams Hematology, 10e Chapter 59: the Porphyrias John D. Inherited mutations inside the genes encoding the eight dedicated steps in heme synthesis have been recognized in each of the porphyrias. Porphyrias can be categorised as both hepatic or erythropoietic, depending on the principal site of preliminary accumulation of extra pathway intermediates. Erythropoietic porphyrias are characterized by childhood onset and a usually stable clinical course. Hepatic porphyrias almost always develop throughout grownup life, and are more variable because of a quantity of influences of medication, hormones, and dietary components on the heme biosynthetic pathway in the liver. The 4 acute porphyrias are associated with neurologic manifestations that usually happen as acute attacks. Disease expression is highly variable in the acute porphyrias, and the good majority of people who inherit mutations of these enzymes stay latent by way of all or most of their lives. Attacks are precipitated by components that improve hepatic heme synthesis, together with sure medicine, sex steroid hormones and their metabolites, and restriction of dietary energy and carbohydrate. Cutaneous porphyrias are associated with both blistering pores and skin lesions or, in the protoporphyrias, with acute nonblistering photosensitivity. Hemolytic anemia is common, and extreme cases could also be transfusion dependent, and will even current in utero with fetal hydrops. Hematopoietic stem cell transplantation in early childhood is the best remedy. An uncommon but probably life-threatening complication is protoporphyric liver failure, which is a result of the cholestatic effects of protoporphyrin, and may require liver transplantation. Sequential or combined hematopoietic stem cell transplantation can scale back the continued contribution of porphyrins from the pink cell compartment and forestall recurrent liver failure in the transplanted liver. Characteristic patterns of these metabolic intermediates in plasma, erythrocytes, urine, and feces are the idea for diagnostic screening exams and extra comprehensive biochemical characterization. Porphyrias are classified as either erythropoietic or hepatic, depending on the principal web site of accumulation of pathway intermediates. Table 59�2 summarizes the most important medical and laboratory options of the porphyrias. Enzymes and intermediates within the heme biosynthetic pathway and the kind of porphyria related to a deficiency of each enzyme (indicated by �). Terms of Use � Privacy Policy � Notice � Accessibility Enzymes and intermediates within the heme biosynthetic pathway and the kind of porphyria associated with a deficiency of each Countway Medical Library enzyme (indicated by �). Terms of Use � Privacy Policy � Notice � Accessibility aPorphyrin levels regular or barely increased. This case was a 33-year-old man with photosensitivity since age 3 months, anemia, splenomegaly, red-wine-colored urine because of a pigment resembling hematoporphyrin, and brown-colored bones at post-mortem. Using out there strategies, their urine was also was shown to comprise a substance related to hematoporphyrin. Anderson Hans G�nther6 revealed a monograph on porphyrins in 1911 and classified porphyrias into four groups: (a) people who have an acute onset without5 / 51 �2021 McGraw Hill. Terms of Use � Privacy Policy � Notice � Accessibility affiliation with drug ingestion; (b) these which may be brought on by sulfonal or trional; (c) hematoporphyria congenita; and (d) persistent hematoporphyria.
Buy discount rizact 10 mg
HbE/-thalassemia shows a remarkable variability in medical expression joint pain treatment options purchase 5mg rizact fast delivery,259�263 ranging from a mild type of thalassemia intermedia to a transfusion-dependent situation clinically indistinguishable from -thalassemia main pain treatment shingles cheap rizact 5 mg fast delivery. Large tumor plenty composed of extramedullary erythropoietic tissue might trigger a wide selection of compression syndromes pain management for arthritis dogs buy rizact 10 mg cheap, including a medical picture that carefully mimics a cerebral tumor. In the milder forms, the main complications are progressive hypersplenism, organ injury as a end result of progressive iron loading from an increased fee of absorption, extramedullary erythropoietic tumor masses, bone disease, and an infection. Usually no HbA is current as a result of the 0-thalassemia is particularly frequent within the elements of the world the place HbE is discovered. The advanced interactions between genetic factors,257,258 variations in adaptation to anemia, significantly in adolescence (see "Pathophysiology" above), and the surroundings, notably proneness to malarial infection, underlie the widely differing and unstable phenotypes of sufferers with HbE/thalassemia. The blood film exhibits severe thalassemic modifications as described above in the Laboratory features section, with massive numbers of nucleated pink cells. Usually no HbA, HbA2, or HbF is current as a end result of no -globin chains are synthesized, though uncommon instances that appear to result from interaction of 0-thalassemia with a extreme nondeletion type of +-thalassemia may show small amounts of HbA. Hemoglobin H Disease Depending on their underlying genotype and other modifiers, these people may have a varying diploma of anemia. Incubation of the pink cells with sensible cresyl blue ends in a number of ragged inclusion bodies in virtually all cells resembling golf balls. These our bodies form because of precipitation of HbH in vitro as a end result of reduction�oxidation motion of the dye. These our bodies are fashioned by in vitro precipitation of the unstable HbH molecule and are seen solely after splenectomy. Note hypochromic pink cells, anisocytosis, goal cells, poikilocytes, including teardrop-shaped purple cells. Note discount in poikilocytes and frequency of target cells, a change according to HbH disease and enhanced by postsplenectomy results. A nucleated purple cell is in this area, reflecting an increase in their prevalence within the blood after splenectomy. An occasional cell with HbH inclusion our bodies may appear after incubation with good cresyl blue. Globinsynthesis studies show a deficit of -globin chain production, with an -chain�to�-chain production ratio of roughly 0. Homozygous State for Nondeletion Types of -Thalassemia the homozygous state for nondeletion types of -thalassemia involving the dominant (2) globin gene causes a more severe deficit of -globin chains than do the deletion forms of +-thalassemia. In the homozygous state for HbConstant Spring or different chain-termination mutations, the blood image shows gentle thalassemic adjustments with normalsize pink cells235,236 with an associated reasonably extreme hemolytic anemia. The homozygous states for the opposite nondeletion forms of +-thalassemia are associated with HbH disease. The Hb sample is regular except for the presence of small quantities (approximately 0. The latter may be noticed on alkaline starch-gel electrophoresis as a faint band migrating between HbA2 and the origin. Starch gel electrophoresis of 1, 2, regular adult; 3, 4, compound heterozygotes for HbConstant Spring and 0-thalassemia with HbH illness; 5, normal grownup; and 6, compound heterozygote for 0-thalassemia and HbConstant Spring. Given the principally normal hematologic image, this genotype usually is referred to as "silent provider. Terms of Use � Privacy Policy � Notice � Accessibility for evaluating the imply �to� ratio with that of regular control subjects. Individuals who inherit variants of this kind and an 0-thalassemia determinant have a type of HbH illness during which the Hb consists of the chain variant Hb and HbH. To obtain these goals, the approaches embody the judicious use of transfusions, the suitable use of iron-chelation remedy, common monitoring for and prompt treatment of complications, and general good supportive care, including psychosocial help. Most sufferers within the developed world are handled at complete thalassemia facilities; a therapy strategy is strongly recommended because it incorporates all of the above in an individually tailor-made programmatic method. Individuals with thalassemia minor hardly ever, if ever, require the treatments described herein. Although the administration mentioned on this part applies principally to sufferers with thalassemia main, it additionally applies to those with thalassemia intermedia as they transfer toward transfusion dependence. The decision to initiate a daily transfusion regimen is comparatively easy for sufferers with homozygous -thalassemia or -thalassemia. However, in patients with medical thalassemia intermedia, it might be more complex and involve patient and family choice, diploma of anemia and bone modifications associated to ineffective erythropoiesis, degree of extramedullary hematopoiesis, and issues such as spinal wire compression, nonhealing leg ulcers, or progressive signs related to anemia. Some patients with thalassemia intermedia may only require periodic transfusions at times of stress, corresponding to severe infections, surgical procedure, or pregnancy, whereas others might show a progressive course with extra issues and show a profit from beginning common transfusions to stop progression. This permits children with -thalassemia to develop and develop normally, with out the distressing skeletal issues of thalassemia occurring. In the severest forms of thalassemia, transfusions are generally begun in the first or second 12 months of life, and continued monthly. To avoid transfusion reactions, washed, filtered, or frozen purple cells should be used so that nearly all of the white cells and plasma-protein components are with -thalassemia to grow and develop normally, with out the distressing skeletal problems of thalassemia occurring. It is recommended to have a restricted prolonged phenotype of the recipient purple cells, generally together with the C, D, E, and Kell pink cell antigens (Chap. To avoid transfusion reactions, washed, filtered, or frozen purple cells should be used so that almost all of the white cells and plasma-protein elements are removed (Chap. The quantity of packed pink blood cells transfused at every visit is mostly 15 mL/kg in youngsters, to increase to a full unit when the child weighs approximately 15 kg. The interval between transfusions may be shortened as a substitute, until the kid reaches a weight of approximately 36�40 kg, when a second unit may be added. Transfusion reactions vary from nonhemolytic complications like febrile reactions, allergic reactions together with urticarial reactions, and rare anaphylaxis, to hemolytic complications like acute and delayed hemolysis. The improvement of antibodies, both autoantibodies and alloantibodies are well known in thalassemia sufferers. Alloantibodies make it tougher to crossmatch blood for sufferers, although the incidence of growth of latest alloantibodies is declining with the institution of extra standard extended typing. To decrease the development of hypersplenism and the increased risk of iron overload, as well as to reduce the chance of spontaneous or traumatic rupture of a large spleen, the spleen was surgically eliminated. Ongoing ineffective erythropoiesis, extramedullary hematopoiesis, and enlargement of the spleen with elevated transfusion requirements occurs commonly in patients maintained at a lower Hb degree. The rationale proposed for shifting away from splenectomy is that the lack of splenic clearance of deformed and fragmented cells, together with the ensuing thrombocytosis, will increase the risk of developing vascular disease, notably pulmonary hypertension. In sufferers transfused on an irregular basis, this would be a threat issue, and we do see an increased prevalence of vascular disease, including pulmonary hypertension and silent cerebral infarcts within the thalassemia intermedia population. Patients must be positioned on prophylactic oral penicillin postsplenectomy, and any febrile episode must be handled with warning, with assessment and prompt administration of antibiotics as indicated. Patients with thalassemia and iron overload are predisposed to certain infections apart from those anticipated on account of splenectomy alone. Presentation with belly pain, diarrhea, and vomiting should always suggest an an infection with a member of the Yersinia class of bacteria, which is an iron-avid organism. In the quick term, patients may have reactions as discussed in "Transfusion" part on previous page, but long-term problems are more worrisome.

Discount 5 mg rizact mastercard
Myelodysplastic syndrome difficult with inflammatory intestinal ulcers: significance of trisomy eight oriental pain treatment center brentwood safe 10 mg rizact. Risk factor evaluation in myelodysplastic syndrome sufferers with del(20q): prognosis revisited acute chest pain treatment guidelines purchase 10 mg rizact otc. Identification of del(20q) in a subset of patients recognized with idiopathic thrombocytopenic purpura pain treatment consultants of wny order 10 mg rizact visa. Characterization of chromosome arm 20q abnormalities in myeloid malignancies using genome-wide single nucleotide polymorphism array analysis. Deletion of Asxl1 ends in myelodysplasia and extreme developmental defects in vivo. Investigation of 305 patients with myelodysplastic syndromes and 20q deletion for associated cytogenetic and molecular genetic lesions and their prognostic impact. Myelodysplastic syndrome with isolated deletion of chromosome 20q: an indolent illness with minimal morphological dysplasia and frequent thrombocytopenic presentation. Loss of the Y chromosome: an age-related or clonal phenomenon in acute myelogenous leukemia/myelodysplastic syndrome Complex, not monosomal, karyotype is the cytogenetic marker of poorest prognosis in patients with primary myelodysplastic syndrome. Monosomal karyotype in myelodysplastic syndromes, with or without monosomy 7 or 5, is prognostically worse than an in any other case advanced karyotype. Driver somatic mutations establish distinct illness entities within myeloid neoplasms with myelodysplasia. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. Physiologic expression of Sf3b1(K700E) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Tet2 loss leads to elevated hematopoietic stem cell self-renewal and myeloid transformation. Deletion of Tet2 in mice results in dysregulated hematopoietic stem cells and subsequent growth of myeloid malignancies. Oncogenic isocitrate dehydrogenase mutations: mechanisms, fashions, and scientific opportunities. Oncometabolite 2-hydroxyglutarate is a aggressive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Inactivation of polycomb repressive advanced 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Somatic mutations predict poor end result in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. Somatic mutations predict poor end result in patients with myelodysplastic syndrome after hematopoietic Access Provided by: stem-cell transplantation. Genetic abnormalities in myelodysplasia and secondary acute myeloid leukemia: impression on end result of stem cell transplantation. Mutations of e3 ubiquitin ligase cbl members of the family represent a novel frequent pathogenic lesion in myeloid malignancies. Recurrent mutations in a number of parts of the cohesin complicated in myeloid neoplasms. Increased circulating colony-stimulating factor-1 in patients with preleukemia, leukemia, and Countway Medical Library 256. Increased circulating colony-stimulating factor-1 in sufferers with preleukemia, leukemia, and lymphoid malignancies. Oligoclonal T cell enlargement in myelodysplastic syndrome: evidence for an autoimmune process. Loss of T-lymphocyte clonal dominance in patients with myelodysplastic syndrome conscious of immunosuppression. Somatic mutations establish a sub-group of aplastic anemia patients that progress to myelodysplastic syndrome. Common troublesome symptoms and their impression on quality of life in patients with Countway Medical Library 273. Significance of fetal hemoglobin-containing erythroblasts (F blasts) and the F blast/F cell ratio in myelodysplastic syndromes. An acute leukaemia augured before clinical indicators by blood group antigen abnormalities and low ranges of A and H blood group transferase activities in erythrocytes. S1 nuclease analysis of alpha-globin gene expression in preleukemic patients with acquired hemoglobin H illness after switch to mouse erythroleukemia cells. Acquired alpha-thalassemia in preleukemia is as a result of of decreased expression of all four alpha-globin genes. Clinical and prognostic significance of monocyte count, degree of blastic infiltration, and ring sideroblasts. The preleukemic syndrome: medical and laboratory features, pure course, and administration. Clinical and prognostic significance Countway Medical Library of monocyte depend, Access Provided by: diploma of blastic infiltration, and ring sideroblasts. Pseudo Pelger-Huet anomaly in myelodysplastic syndrome: hyposegmented apoptotic neutrophil Myeloid surface antigen abnormalities in myelodysplasia: relation to prognosis and modification by 13-cis retinoic acid. Quantitative cytochemistry of blood neutrophils in myelodysplastic syndromes and persistent granulocytic leukaemia. Deficiency of neutrophilic granule membrane glycoproteins within the myelodysplastic syndromes: a typical deficiency in 216 patients studied by the Cancer and Leukemia Group B. Flow cytometric assay for the analysis of phagocytosis and oxidative burst of polymorphonuclear leukocytes and monocytes in myelodysplastic disorders. Actin polymerization in neutrophils from sufferers affected by myelodysplastic syndromes-a circulate cytometric research. The myelodysplastic syndromes-a study of haemostatic operate and platelet ultrastructure. Studies of T-lymphocytes in preleukemic problems and acute nonlymphocytic leukemia: in Countway Medical Library 310. Deficiency in Epstein-Barr virus receptors on B-lymphocytes of preleukemia patients. Studies of T-lymphocytes in preleukemic issues and acute nonlymphocytic leukemia: in vitro radiosensitivity, mitogenic responsiveness, colony formation, and enumeration of lymphocytic subpopulations. Cytogenetic evidence for involvement of B lymphocytes in acquired idiopathic sideroblastic anemias. The medical significance of activated lymphocytes in patients with myelodysplastic syndromes: a single centre research of 131 patients.
Generic rizact 5 mg overnight delivery
The therapeutic target is a serum ferritin close to 15 mcg/L pain medication for shingles pain generic 5 mg rizact with mastercard, which is close to pain & depression treatment order rizact 5mg free shipping the decrease restrict of regular and related to tissue iron depletion sickle cell anemia pain treatment guidelines order rizact 10mg without prescription, but normally not anemia. Treatment is also guided by plasma (or serum) porphyrin levels, that are more convenient to measure repeatedly than urine porphyrins, and fall more slowly than the serum ferritin. Plasma porphyrins normally decline from preliminary levels of 10�25 mcg/dL throughout treatment, to under the higher restrict of regular (~1 mcg/dL) inside weeks after phlebotomies are completed. However, relapses could occur, especially in patients who resume use of alcohol, and are handled by one other course of phlebotomies. It can also be advisable to comply with porphyrin levels and reinstitute phlebotomies promptly if porphyrin ranges begin to rise. Liver imaging and a serum -fetoprotein dedication ought to be repeated as screening for hepatocellular carcinoma. A prospective comparative examine discovered that point to biochemical remission with low-dose hydroxychloroquine was comparable to that with phlebotomy. Most doubtless, these drugs colocalize with extra porphyrins in lysosomes and other intracellular organelles and promote their release by a course of that involves transient cell harm. Erythropoietin administration can right anemia, mobilize iron, and help phlebotomy in many cases. Levels of plasma porphyrins are often particularly excessive in these patients, and should be assessed previous to surgical procedure, because there could also be some risk of pores and skin and peritoneal burns from publicity to operating room lights. Retrovirus-mediated gene switch can appropriate porphyria in cell lines from patients with this illness, which means that gene remedy may be applicable sooner or later. Studien uber die entstehung der roten harnpigmente (uroporphyrin und porphobilin) bein der akuten porphyrie aus iher farblosen vorstufe (porphobilinogen). Heme biosynthesis in intermittent acute porphyria: decreased hepatic conversion of porphobilinogen to porphyrins and elevated delta-aminolevulinic acid synthetase activity. Repression of the overproduction of porphyrin precursors in acute intermittent porphyria by intravenous infusions of hematin. Heme biosynthesis in intermittent acute porphyria: decreased hepatic conversion of porphobilinogen to Countway Medical Library porphyrins and increased delta-aminolevulinic acid synthetase exercise. Intramitochondrial localization of delta-aminolaevulate synthetase and ferrochelatase in rat liver. Expression of delta-aminolevulinate synthase in avian cells: separate genes encode erythroid-specific and nonspecific isozymes. Regulation of rat hepatic delta-aminolevulinic acid synthetase and heme oxygenase actions: evidence for control by heme and in opposition to mediation by prosthetic iron. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory components: a potential regulatory hyperlink between nuclear and mitochondrial gene expression in organelle biogenesis. Hepatic nuclear factor 3 and nuclear factor 1 regulate 5-aminolevulinate synthase gene expression and are concerned in insulin repression. Distinctive responses of the erythroid-specific and the nonspecific/ 51 Page 34, John D. Hepatic nuclear factor three and nuclear issue 1 regulate 5-aminolevulinate synthase gene expression and Countway Medical Library are concerned in insulin repression. Pyridoxine refractory X-linked sideroblastic anemia caused by a point mutation within the erythroid 5aminolevulinate synthase gene. The position of zinc with particular reference to the essential thiol teams in delta-aminolevulinic acid dehydratase of bovine liver. Profound inhibition of delta-aminolevulinic acid dehydratase exercise by succinylacetone. Human delta-aminolevulinate dehydratase: chromosomal localization to 9q34 by in situ hybridization. The biosynthesis of 5-aminolevulinic acid and its transformation into coproporphyrinogen in animals and bacteria. Reconstitution of the holoenzyme form of Escherichia coli porphobilinogen deaminase from 50. Reconstitution of the holoenzyme type of Escherichia coli porphobilinogen deaminase from apoenzyme with porphobilinogen and preuroporphyrinogen: a research using circular dichroism spectroscopy. Regional gene task of human porphobilinogen deaminase and esterase A4 to chromosome 11q23 results in 11qter. Alternative transcription and splicing of the human porphobilinogen deaminase gene outcome either in tissue-specific or in housekeeping expression. Molecular cloning and full primary sequence of human erythrocyte porphobilinogen deaminase. Cis - and trans -acting components involved in the regulation of the erythroid promoter of the human porphobilinogen deaminase gene. A level mutation G-A in exon 12 of the porphobilinogen deaminase gene ends in exon skipping and is liable for acute intermittent porphyria. A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda. Interaction between iron metabolism and a pair of,3,7,8-tetrachlorodibenzo-p-dioxin in mice with variants of the Ahr gene: a hepatic oxidative mechanism. Assignment of the gene for uroporphyrinogen decarboxylase to human chromosome 1 by somatic cell hybridization and particular enzyme immunoassay. Localization of the human coproporphyrinogen oxidase gene to chromosome band 3q12. Proteomic mapping of mitochondria in residing cells by way of spatially restricted enzymatic tagging. Mouse coproporphyrinogen oxidase is a copper-containing enzyme: expression in Escherichia coli and site-directed mutagenesis. Cloning of a coproporphyrinogen oxidase promoter regulatory factor binding protein. Multiple mechanisms for the regulation of haem synthesis during erythroid cell differentiation. Identification of sequences required for the import of human protoporphyrinogen oxidase to mitochondria. A pi-helix switch selective for porphyrin deprotonation and product launch in human ferrochelatase. Some studies of the comparative biology of human and bovine porphyria erythropoietica. Complete suppression of the symptoms of congenital erythropoietic porphyria by long-term treatment with high-level transfusions. Successful match-unrelated donor bone marrow transplantation for congenital erythropoietic porphyria (Gunther disease). Congenital erythropoietic porphyria: prolonged high-level expression and correction of the heme biosynthetic defect by retroviral-mediated gene switch into porphyric and erythroid cells. Terms of Use � Privacy Policy � Notice � AccessibilityActa Dermatovenerol Alp Pannonica Adriat. The molecular defect of ferrochelatase in a patient with erythropoietic protoporphyria.
References
- Gosselink ATM, Blanksma PK, Crijns H, et al. Left ventricular beat-to-beat performance in atrial fibrillation: contribution of Frank-Starling mechanism after short rather than long RR intervals. J Am Coll Cardiol 1995;26:1516-1521.
- Ganesan S, Faris AN, Comstock AT, et al. Elastase/LPS-exposed mice exhibit impaired innate immune responses to bacterial challenge: role of scavenger receptor A. Am J Pathol 2012; 180: 61-72.
- Zhang ZX, Milich DR, Peterson DL, et al. Interferon alfa treatment induces delayed CD4 proliferative responses to the hepatitis C virus nonstructural protein 3 regardless of the outcome of therapy. J Infect Dis. 1997;175:1294-1301.
- Morrow DA, Cannon CP, Rifai N, et al: Ability of minor elevations of troponins I and T to predict benefit from an early invasive strategy in patients with unstable angina and non-ST elevation myocardial infarction: Results from a randomized trial. JAMA 2001;286:2405-2412.
- Parekh DJ, Weinberg JM, Ercole B, et al. Tolerance of the human kidney to isolated controlled ischemia. J Am Soc Nephrol. 2013;24:506-517.